Ependymomas are typically slow-growing tumors of the central nervous system. They can occur at any age, but are more common in children and adolescents. While some ependymomas remain entirely asymptomatic, others can cause severe neurological issues, particularly when they obstruct the flow of cerebrospinal fluid. The optimal treatment involves complete surgical removal, often supplemented with radiation or chemotherapy in many cases to reduce the risk of recurrence.
Who is affected by ependymomas?
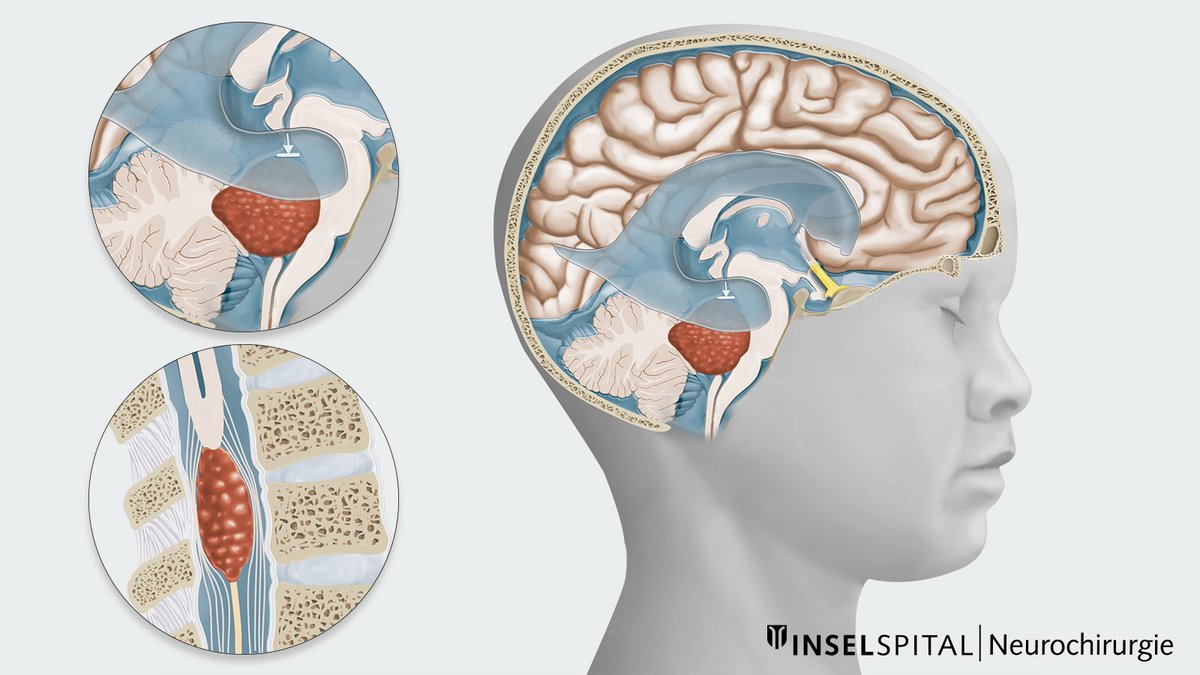
Ependymomas can occur in any age group and throughout the entire range of the ventricular system and spinal cord. The location of the tumor and the age of onset vary depending on the type of ependymoma.
In general:
- Ependymomas are more commonly diagnosed in childhood and adolescence, with about 25% occurring in children under 3 years of age.
- Ependymomas account for approximately 10% of all pediatric brain tumors.
- Nearly two-thirds of ependymomas located in the skull occur in children, while tumors in the spinal cord are almost exclusively diagnosed in adults or adolescents.
- Besides the common occurrence in children, ependymomas are more frequently seen in adults between the ages of 30 and 60.
What are the different types of ependymomas?
The group of ependymomas is very heterogeneous and includes both benign, slow-growing tumors and aggressive, fast-growing tumors. Additionally, there are various molecular subgroups associated with different prognoses.
Traditionally, ependymomas are classified according to their histology into WHO grades 1–3. However, it has been shown that risk assessment based on molecular subgroups, tumor localization, and patient age is more suitable. There is still insufficient data available to assign individual molecular subgroups to WHO grades *.
According to the new WHO classification, ependymomas are now classified into *:
Supratentorial ependymoma
Localization cerebrum, WHO grade 2–3, typically in younger children
Molecular subgroups:
- ST ependymoma with ZFTA fusion (formerly RELA fusion)
- ST ependymoma with YAP1 fusion
- Other rare fusions
Infratentorial ependymoma
Posterior fossa localization, WHO grade 2–3, more common in older children and adults
Molecular subgroups:
- PFA ependymoma (PF type A)
- PFB ependymoma (PF type B)
Spinal ependymoma
MYCN amplification, WHO grade 2–3
Myxopapillary ependymoma
formerly WHO grade 1, now WHO grade 2, special type of spinal ependymoma, mostly in older children and young adults
Subependymoma
WHO grade 1
What are the symptoms of an ependymoma?
Depending on their growth rate, ependymomas growing in the brain can lead to a blockage of the cerebrospinal fluid in the ventricular system and thus to an occlusive hydrocephalus. This results in an increase in pressure in the ventricular system, which can lead to headaches, nausea, vomiting, unsteady gait and clouding of consciousness or even coma.
Ependymomas can also lead to facial, pharyngeal and ocular muscle deficits due to the involvement of cranial nerves.
In infants, an ependymoma can manifest itself in an enlargement of the skull circumference (macrocephaly) and a bulging and tense fontanel due to the unsealed cranial sutures. This often occurs before the child shows other symptoms such as developmental delays, nausea or vomiting.
Patients with intramedullary ependymomas primarily complain of local, non-radiating back pain. Furthermore, around half of patients have sensory disturbances and strength deficits at the time of diagnosis.
A spinal movement disorder (ataxia) with tumor growth in the neck and neck area as well as urinary bladder or rectal dysfunction, on the other hand, occur rarely.
How is an ependymoma diagnosed?
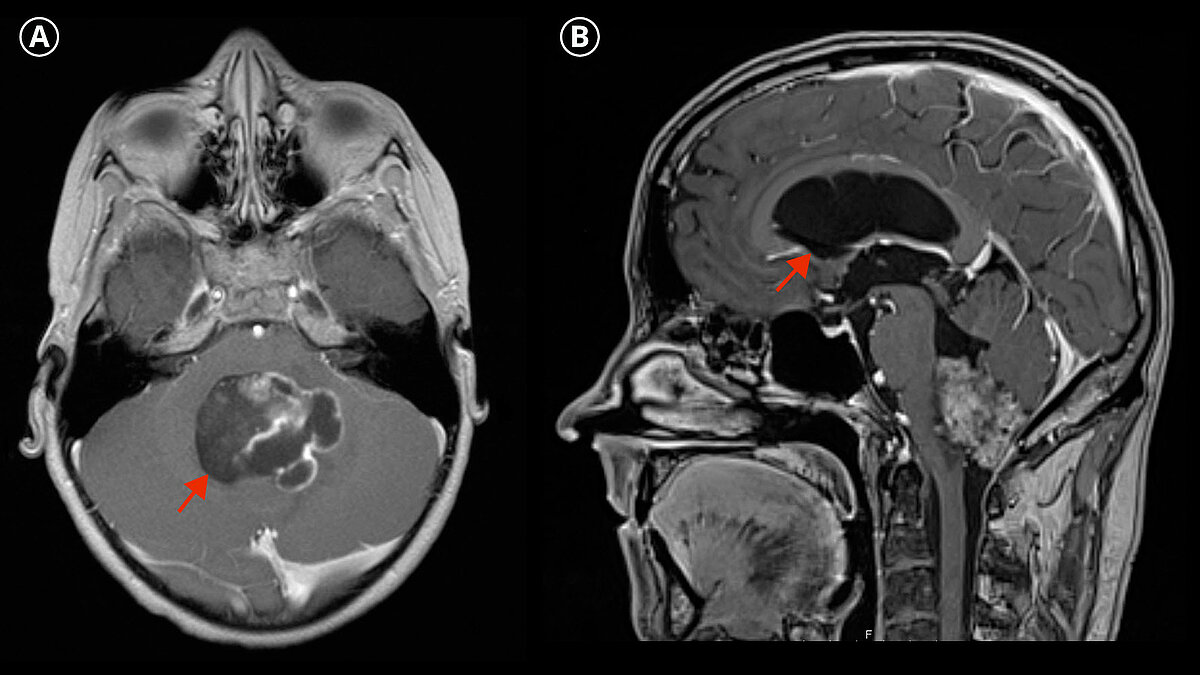
In imaging diagnostics, ependymomas typically appear as a heterogeneous mass with solid or necrotic or cystic parts, whereby hemorrhages and calcifications are not uncommon.
The imaging diagnosis is made by magnetic resonance imaging (MRI).
To exclude possible metastases, an imaging examination of the entire central nervous system including the spinal cord must always be performed.
In up to 10 % of patients, tumor cells may seed in the cerebrospinal fluid system. This is referred to as drop metastases. Therefore, an additional lumbar puncture must be performed for cytological detection of tumor cells.
How is an ependymoma treated?
The treatment of ependymoma depends on various factors, including the patient's age, tumor location, tumor grade and molecular characteristics. In general, treatment includes the following steps *, *:
Surgical resection
Maximum safe surgical removal of the tumor is the main goal of treatment and has a strong impact on prognosis. In special subgroups of ependymomas (PFB and YAP fusion, spinal ependymomas WHO grade 2), resection without radiotherapy may even be possible due to the good prognosis. In the case of recurrence, another resection surgery should be discussed.
If a perfect cerebrospinal fluid drainage cannot be restored by a maximum and safe tumor resection, an additional operation is required for an alternative drainage of the cerebrospinal fluid. This can be achieved by means of an endoscopic third ventriculostomy or a ventriculoperitoneal shunt, for example.
Radiotherapy
Most ependymomas require adjuvant radiotherapy to reduce the risk of recurrence. If the tumor has already metastasized, a more intensive dose of radiation is necessary. However, radiotherapy is not always possible due to the young age of the patients. Chemotherapy can therefore be used as an alternative, particularly in children under 12 months of age.
Chemotherapy
The role of chemotherapy in ependymomas is controversially discussed. In some cases, such as when the patient is too young for radiotherapy, it can be used as a delaying step.
Long-term aftercare
Due to the risk of late recurrence and long-term consequences of therapy, long-term follow-up care is important in order to detect and treat tumor regrowth at an early stage.
The specific treatment strategy is determined individually for each patient and can vary depending on the individual characteristics of the tumor and the response to therapy. Interdisciplinary care is essential for optimum treatment.
What is the complication rate of the therapy?
In general, the complication rate of both tumor surgery and radiotherapy is influenced by many factors. The size and location of the tumor, its accessibility as well as the clinical condition and age of the patient should be emphasized here.
Depending on the location of the tumor, there is a certain risk of damage to adjacent structures during surgery. The posterior fossa syndrome with cerebellar mutism following infratentorial surgery is particularly worth mentioning here. It occurs much more frequently in children. This syndrome often leads to loss of speech in combination with other symptoms, such as emotional instability, reduced muscle tone and movement disorders.
There is also a risk of spreading tumor cells during the operation, which can lead to drop metastases.
In order to remove the tumor completely and keep the risk of complications as low as possible, we at Inselspital use special innovative procedures such as intraoperative imaging with MRI, tumor visualization with fluorescent dyes, neuronavigation and specialized intraoperative neuromonitoring.
What is the prognosis for an ependymoma?
The prognosis depends on various factors such as tumor location, patient age, tumor subgroup and the extent of tumor removal.
Besides the histological grading according to the WHO classification, complete surgical removal is the most important prognostic factor.
The molecular subtype of the tumor is also important. The prognosis varies greatly within the different subtypes. For example, supratentorial ependymomas with ZFTA fusion and YAP fusion show a good outcome. In the case of infratentorial ependymomas, PFA ependymomas usually occur in younger children and are associated with a poor prognosis. PFB ependymomas are more common in older children and adults and show a more favorable prognosis with cure by surgical resection alone.
The 5-year survival rate in children therefore varies between 50 % and 75 %, although mortality is significantly higher if the tumor has already recurred. Supportive radiotherapy can reduce the recurrence rate, especially in cases of incomplete tumor removal.
-
Lutz K, Jünger ST, Messing-Jünger M. Essential Management of Pediatric Brain Tumors. Children (Basel). 2022 Apr 2;9(4):498. doi: 10.3390/children9040498.
-
Central Nervous System Tumours, WHO Classification of Tumours, 5th Edition, Volume 6, 2021, ISBN-13-978-92-832-4508-7
-
Rudà R, Reifenberger G, Frappaz D, Pfister SM, Laprie A, Santarius T, Roth P, Tonn JC, Soffietti R, Weller M, Moyal EC. EANO guidelines for the diagnosis and treatment of ependymal tumors. Neuro Oncol. 2018 Mar 27;20(4):445-456. doi: 10.1093/neuonc/nox166.
Further reading
- Berlit P. Basiswissen Neurologie. 6. Auflage. Berlin Heidelberg: Springer-Verlag; 2014.
- Chamberlain M. Intracranial ependymoma. In: International Neurology. 2nd Edition. Chichester: John Wiley & Sons, Ltd; 2016.
- Hacke W. Neurologie. 14. Auflage. Berlin Heidelberg: Springer-Verlag; 2016.
- Massimino M, Barretta F, Modena P, Witt H, Minasi S, Pfister SM, Pajtler KW, Antonelli M, Gandola L, Luisa Garrè M, Bertin D, Mastronuzzi A, Mascarin M, Quaglietta L, Viscardi E, Sardi I, Ruggiero A, Pollo B, Buccoliero A, Boschetti L, Schiavello E, Chiapparini L, Erbetta A, Morra I, Gessi M, Donofrio V, Patriarca C, Giangaspero F, Johann P, Buttarelli FR. Second series by the Italian Association of Pediatric Hematology and Oncology of children and adolescents with intracranial ependymoma: an integrated molecular and clinical characterization with a long-term follow-up. Neuro Oncol. 2021 May 5;23(5):848-857. doi: 10.1093/neuonc/noaa257.
- Merchant TE, Bendel AE, Sabin ND, Burger PC, Shaw DW, Chang E, Wu S, Zhou T, Eisenstat DD, Foreman NK, Fuller CE, Anderson ET, Hukin J, Lau CC, Pollack IF, Laningham FH, Lustig RH, Armstrong FD, Handler MH, Williams-Hughes C, Kessel S, Kocak M, Ellison DW, Ramaswamy V. Conformal Radiation Therapy for Pediatric Ependymoma, Chemotherapy for Incompletely Resected Ependymoma, and Observation for Completely Resected, Supratentorial Ependymoma. J Clin Oncol. 2019 Apr 20;37(12):974-983. doi: 10.1200/JCO.18.01765.
- Merchant TE, Li C, Xiong X, Kun LE, Boop FA, Sanford RA. Conformal radiotherapy after surgery for paediatric ependymoma: a prospective study. Lancet Oncol. 2009 Mar;10(3):258-66. doi: 10.1016/s1470-2045(08)70342-5
- Upadhyaya SA, Robinson GW, Onar-Thomas A, Orr BA, Billups CA, Bowers DC, Bendel AE, Hassall T, Crawford JR, Partap S, Fisher PG, Tatevossian RG, Seah T, Qaddoumi IA, Vinitsky A, Armstrong GT, Sabin ND, Tinkle CL, Klimo P, Indelicato DJ, Boop FA, Merchant TE, Ellison DW, Gajjar A. Molecular grouping and outcomes of young children with newly diagnosed ependymoma treated on the multi-institutional SJYC07 trial. Neuro Oncol. 2019 Oct 9;21(10):1319-1330. doi: 10.1093/neuonc/noz069.
- Pajtler KW, Witt H, Sill M, Jones DT, Hovestadt V, Kratochwil F, Wani K, Tatevossian R, Punchihewa C, Johann P, Reimand J, Warnatz HJ, Ryzhova M, Mack S, Ramaswamy V, Capper D, Schweizer L, Sieber L, Wittmann A, Huang Z, van Sluis P, Volckmann R, Koster J, Versteeg R, Fults D, Toledano H, Avigad S, Hoffman LM, Donson AM, Foreman N, Hewer E, Zitterbart K, Gilbert M, Armstrong TS, Gupta N, Allen JC, Karajannis MA, Zagzag D, Hasselblatt M, Kulozik AE, Witt O, Collins VP, von Hoff K, Rutkowski S, Pietsch T, Bader G, Yaspo ML, von Deimling A, Lichter P, Taylor MD, Gilbertson R, Ellison DW, Aldape K, Korshunov A, Kool M, Pfister SM. Molecular Classification of Ependymal Tumors across All CNS Compartments, Histopathological Grades, and Age Groups. Cancer Cell. 2015 May 11;27(5):728-43. doi: 10.1016/j.ccell.2015.04.002.
- Elsamadicy AA, Koo AB, David WB, Lee V, Zogg CK, Kundishora AJ, Hong CS, DeSpenza T, Reeves BC, Kahle KT, DiLuna M. Comparison of epidemiology, treatments, and outcomes in pediatric versus adult ependymoma. Neurooncol Adv. 2020 Feb 21;2(1):vdaa019. doi: 10.1093/noajnl/vdaa019.
- McGuire CS, Sainani KL, Fisher PG. Incidence patterns for ependymoma: a surveillance, epidemiology, and end results study. J Neurosurg. 2009 Apr;110(4):725-9. doi: 10.3171/2008.9.JNS08117.
- Gerstner ER, Pajtler KW. Ependymoma. Semin Neurol. 2018 Feb;38(1):104-111. doi: 10.1055/s-0038-1636503.
- Taylor MD, Poppleton H, Fuller C, Su X, Liu Y, Jensen P, Magdaleno S, Dalton J, Calabrese C, Board J, Macdonald T, Rutka J, Guha A, Gajjar A, Curran T, Gilbertson RJ. Radial glia cells are candidate stem cells of ependymoma. Cancer Cell. 2005 Oct;8(4):323-35. doi: 10.1016/j.ccr.2005.09.001.