Pilocytic astrocytoma is the most common benign brain tumor in children and adolescents. It is usually curable by microneurosurgical treatment, even if it is located in a difficult-to-operate site in the brain. After complete removal of the tumor, radiation or chemotherapy are not necessary. In addition to information on standard therapy, below you will also find information on special cases, such as the treatment of children with neurofibromatosis.
What is a pilocytic astrocytoma?
Pilocytic astrocytomas are the most common benign brain tumors of children and young adults, and account for approximately 5% of all gliomas *. They arise from astrocytes, which are part of the supporting tissue (glial cells) of the central nervous system and are therefore classified as gliomas. Microscopically, pilocytic astrocytomas have a fibrous "pilocytic" character. Macroscopically, they are often cystic, meaning that pilocytic astrocytomas consist of a mixture of fluid-filled cavities and solid portions. Most pilocytic astrocytomas are slow-growing tumors that do not spread to other parts of the body and have a very good prognosis.
Where in the brain do pilocytic astrocytomas occur?
In children, pilocytic astrocytomas arise frequently from the astrocyte cells of the cerebellum, a region that plays a crucial role in movement and balance. However, pilocytic astrocytomas can also arise in the visual pathway, cerebrum, or brainstem *, *.
How common are pilocytic astrocytomas?
Pilocytic astrocytomas are the most common primary benign brain tumors in children aged 0–14 years. However, they may continue to occur into adulthood and are slightly more common in males than females (1.1:1 ratio). Pilocytic astrocytomas occur in children at a frequency of 0.8 per 100,000 persons per year.
What are the symptoms of pilocytic astrocytoma?
Pilocytic astrocytomas can increase the pressure inside the skull due to their growth and then lead to:
- morning headaches
- nausea and vomiting
- fatigue
In addition, other symptoms may occur depending on the location of the tumor:
- gait and coordination disorders
- double images
- visual disturbances
- increasing somnolence (sleepiness, drowsiness)
- in rare cases epileptic seizures
- movement and speech disorders
- creeping change of mood and personality
Due to the usually very slow growth of pilocytic astrocytomas, symptoms may appear gradually and go unnoticed for a long time.
What is the cause of pilocytic astrocytomas?
The causes for the development of pilocytic astrocytomas are currently the subject of intensive research. In a large proportion of these tumors, a special variant of a gene called BRAF is found, which is involved in the control of cell division *. In pilocytic astrocytomas, the BRAF gene has a mutation that initiates uncontrolled division of the tumor cells. The effect is mediated by the so-called mitogen-activated protein kinase pathway (MAP kinase pathway or MAPK pathway) *. This is also associated with cell senescence genes, which may explain the slow growth and spontaneous regression of pilocytic astrocytomas described in rare cases * *.
How are pilocytic astrocytomas diagnosed?
Imaging techniques play a key role in the diagnosis of pilocytic astrocytomas. Magnetic resonance imaging (MRI) in combination with an intravenous contrast agent is particularly well suited to visualize pilocytic astrocytomas and to differentiate them from the surrounding healthy brain tissue. However, a MRI cannot diagnose a pilocytic astrocytoma with 100% certainty or rule out the possibility that it is a more malignant tumor. This is possible only after tissue sampling and fine-tissue microscopic examination. This process takes several days. Tissue removal is done either as a biopsy or more often right away during surgical removal of the pilocytic astrocytoma.
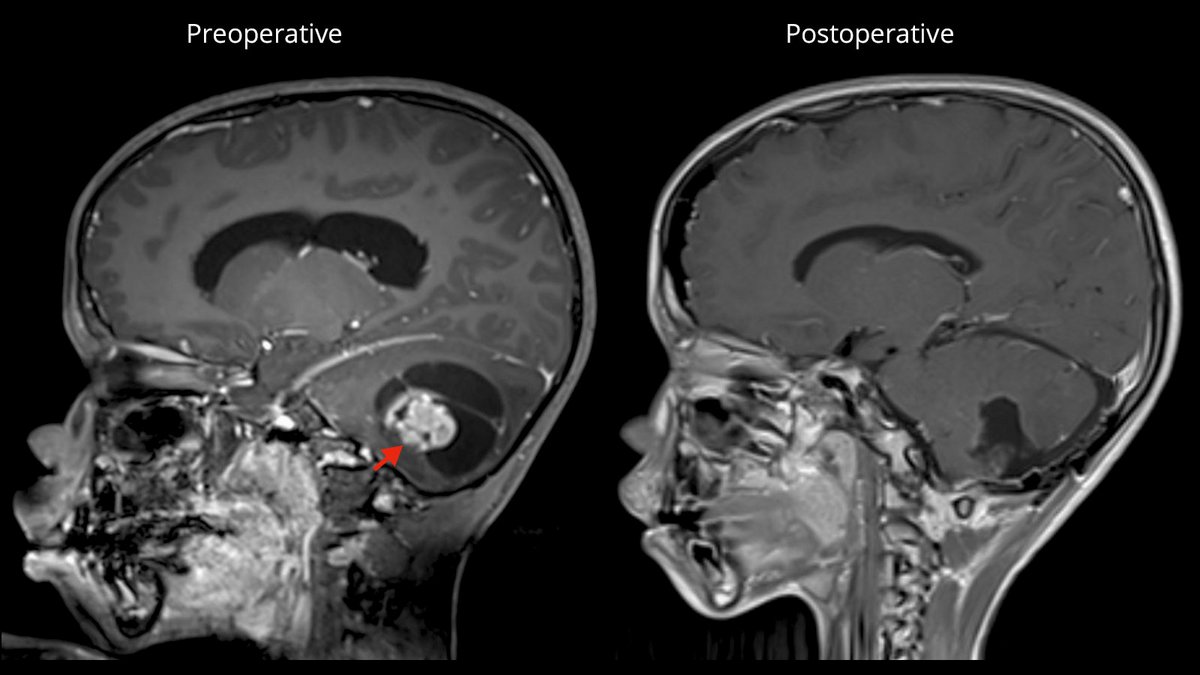
Surgery as the therapy of first choice
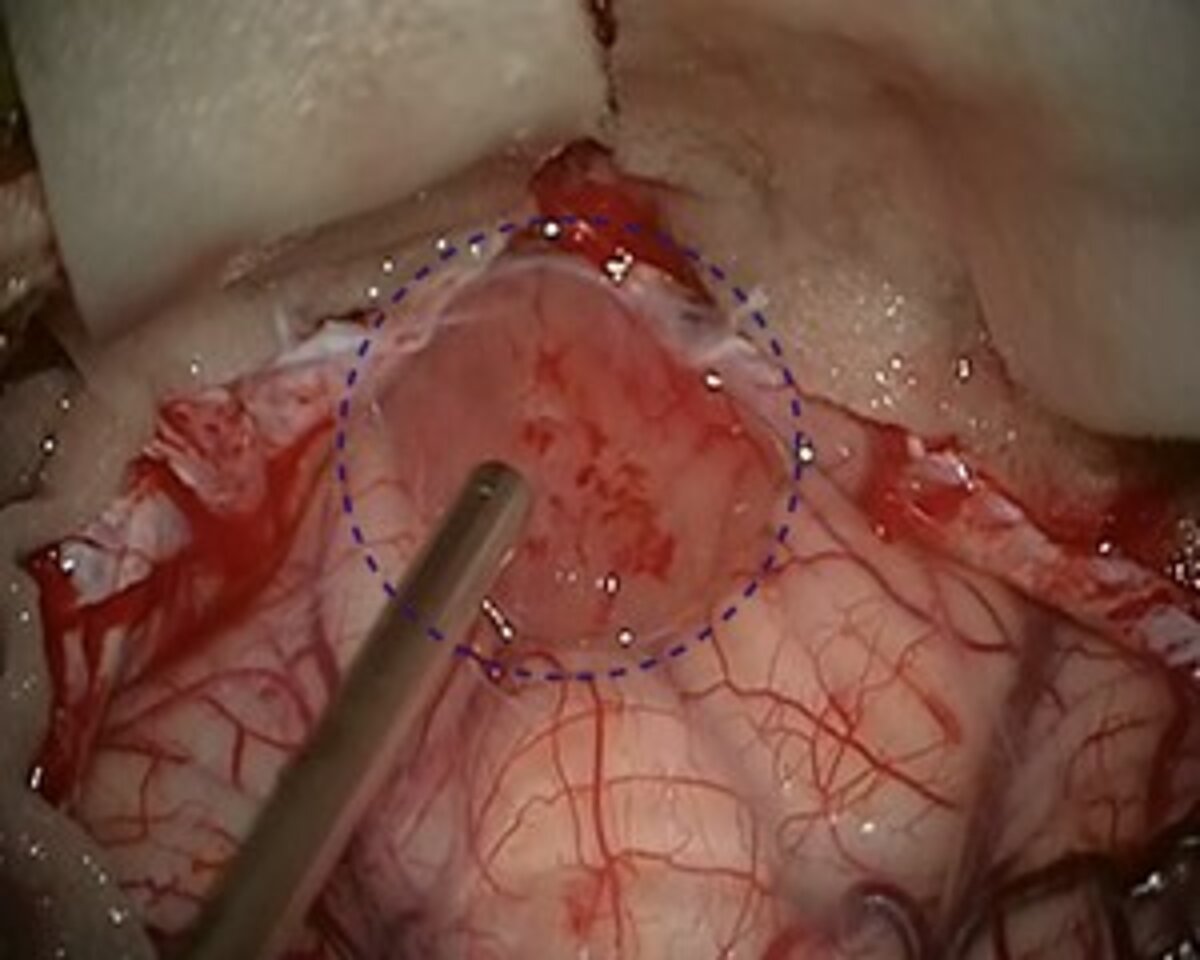
If possible, the first choice treatment is the complete microsurgical removal of the pilocytic astrocytoma. Especially in juveniles, a cure can be achieved after complete removal of the tumor in most cases with a 10-year survival rate of up to 100% and a recurrence rate of only 2–5% *. After complete tumor removal, neither radiation nor chemotherapy is necessary.
In some cases, however, it is not possible to completely remove a pilocytic astrocytoma because it grows into vital structures such as the brain stem. Provided that the tumor has a clear boundary with the surrounding brain tissue, partial removal of the tumor may also be useful in these cases.
Although in the case of residual tumor progression occurs in about half of the patients within 5 years, the 5-year survival is still 90% *, *, *. The complication rate of surgical tumor removal and possible further treatment with radio- and/or chemotherapy is influenced by several factors such as size and location of the tumor, its accessibility, and the clinical condition and age of the patient
Radiotherapy as an addition for residual tumors
If a residual pilocytic astrocytoma remains after surgery and cannot be surgically removed, radiotherapy is an effective option. In recent years, radiosurgery in particular, a very targeted and intensive type of radiation, has become increasingly important. With radiosurgery, good tumor control can be achieved both in the case of a small residual tumor and in the case of a recurrence *, *, *. Alternatively, in the case of residual tumor, one can also wait with further therapy and monitor the patient closely until the residual tumor continues to grow.
Avoidance of radiotherapy in younger children
Radiation in early childhood can lead to cognitive and hormonal problems, as well as vascular changes *, *. For this reason, in younger children under 5–10 years of age, attempts are made to delay radiation and to address any residual tumor after surgery with chemotherapy to spare the growing brain the potentially harmful effect of radiation *, *.
Special case neurofibromatosis type 1
In some cases, pilocytic astrocytomas are due to a hereditary disease, neurofibromatosis type 1 (NF1). In this case, the tumors appear with a different distribution pattern and usually grow even more slowly.
In patients with neurofibromatosis type 1, pilocytic astrocytomas occur most frequently (two-thirds) in the optic nerve or tract, 20% in the brainstem, and only rarely (5%) in the cerebellum *. These tumors exhibit different growth behavior and require adapted therapy. NF1-associated tumors have a better prognosis than sporadic gliomas. There are even reports of spontaneous regression, which is attributed to the aforementioned activation of cellular aging mechanisms *.
Gliomas of the visual pathway
In children with neurofibromatosis type 1, gliomas of the visual pathway are detected in 15–20% of cases, of which, however, only half of these patients ever experience symptoms *. More than half of the tumors of the visual pathway are pilocytic astrocytomas on fine tissue examination *. For gliomas of the visual pathway, no treatment is necessary in most cases *. If treatment is eventually required, it is primarily by chemotherapy.
Special case pilomyxoid astrocytoma
Related to pilocytic astrocytomas are pilomyxoid astrocytomas. While they share many genetic features with pilocytic astrocytomas, they form a distinct subgroup with a more aggressive behavior *. While pilocytic astrocytomas are classified as grade I tumors according to WHO, pilomyxoid astrocytomas are no longer given a grade classification in the new WHO classification of 2016 (formerly grade II) *.
Why you should seek treatment at Inselspital
The top priority of our treatment is maximum tumor removal that is both gentle and preserves function. Depending on the location of the tumor, technical procedures such as neuronavigation and intraoperative neuromonitoring are used for this purpose, which helps the neurosurgeon to work with maximum precision and safety under the operating microscope.
-
Ostrom QT, Gittleman H, Xu J et al. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2009-2013. Neuro Oncol. 2016;18:v1-v75.
-
Fernandez C, Figarella-Branger D, Girard N et al. Pilocytic astrocytomas in children: prognostic factors--a retrospective study of 80 cases. Neurosurgery. 2003;53:544-53; discussion 554.
-
Bond KM, Hughes JD, Porter AL, Orina J, Fang S, Parney IF. Adult Pilocytic Astrocytoma: An Institutional Series and Systematic Literature Review for Extent of Resection and Recurrence. World Neurosurg. 2018;110:276-283.
-
Korshunov A, Meyer J, Capper D et al. Combined molecular analysis of BRAF and IDH1 distinguishes pilocytic astrocytoma from diffuse astrocytoma. Acta Neuropathol. 2009;118:401-405.
-
Tatevossian RG, Lawson AR, Forshew T, Hindley GF, Ellison DW, Sheer D. MAPK pathway activation and the origins of pediatric low-grade astrocytomas. J Cell Physiol. 2010;222:509-514.
-
Jacob K, Quang-Khuong DA, Jones DT et al. Genetic aberrations leading to MAPK pathway activation mediate oncogene-induced senescence in sporadic pilocytic astrocytomas. Clin Cancer Res. 2011;17:4650-4660.
-
Raabe EH, Lim KS, Kim JM et al. BRAF activation induces transformation and then senescence in human neural stem cells: a pilocytic astrocytoma model. Clin Cancer Res. 2011;17:3590-3599.
-
Trifiletti DM, Peach MS, Xu Z, Kersh R, Showalter TN, Sheehan JP. Evaluation of outcomes after stereotactic radiosurgery for pilocytic astrocytoma. J Neurooncol. 2017;134:297-302.
-
Simonova G, Kozubikova P, Liscak R, Novotny J. Leksell Gamma Knife treatment for pilocytic astrocytomas: long-term results. J Neurosurg Pediatr. 2016;18:58-64.
-
Kano H, Niranjan A, Kondziolka D et al. Stereotactic radiosurgery for pilocytic astrocytomas part 2: outcomes in pediatric patients. J Neurooncol. 2009;95:219-229.
-
Ater JL, Zhou T, Holmes E et al. Randomized study of two chemotherapy regimens for treatment of low-grade glioma in young children: a report from the Children’s Oncology Group. J Clin Oncol. 2012;30:2641-2647.
-
Gnekow AK, Walker DA, Kandels D et al. A European randomised controlled trial of the addition of etoposide to standard vincristine and carboplatin induction as part of an 18-month treatment programme for childhood (≤16 years) low grade glioma - A final report. Eur J Cancer. 2017;81:206-225.
-
Merchant TE, Kun LE, Wu S, Xiong X, Sanford RA, Boop FA. Phase II trial of conformal radiation therapy for pediatric low-grade glioma. J Clin Oncol. 2009;27:3598-3604.
-
Merchant TE, Conklin HM, Wu S, Lustig RH, Xiong X. Late effects of conformal radiation therapy for pediatric patients with low-grade glioma: prospective evaluation of cognitive, endocrine, and hearing deficits. J Clin Oncol. 2009;27:3691-3697.
-
Campen CJ, Gutmann DH. Optic Pathway Gliomas in Neurofibromatosis Type 1. J Child Neurol. 2018;33:73-81.
-
Rodriguez FJ, Perry A, Gutmann DH et al. Gliomas in neurofibromatosis type 1: a clinicopathologic study of 100 patients. J Neuropathol Exp Neurol. 2008;67:240-249.
-
Rubin JB, Gutmann DH. Neurofibromatosis type 1 - a model for nervous system tumour formation. Nat Rev Cancer. 2005;5:557-564.
-
Banan R, Hartmann C. The new WHO 2016 classification of brain tumors-what neurosurgeons need to know. Acta Neurochir (Wien). 2017;159:403-418.